

Abstract
Hereditary hemolytic anemia (HHA) are a heterogeneous group of disorders due to germline mutations of the red cell cytoskeleton (e.g. hereditary spherocytosis (HS) and hereditary elliptocytosis/pyropoikilocytosis (HE/HPP)) or enzyme deficiencies (e.g. glucose 6 phosphate dehydrogenase deficiency (G6PD) and pyruvate kinase deficiency (PKD). Routine morphological and biochemical analysis may be inconclusive in neonates due to the physiological nature of erythroid cell maturation and can also be misleading in transfusion-dependent patients. Additionally, there has been increasing awareness of inherited red cell membrane disorders that are not easily identified by routine laboratory approaches. For example, clinically insignificant defects of RBC membrane genes (e.g. alpha LELY and alpha LEPRA in SPTA1), which can be present in the parents without significant hemolysis, may result in compound heterozygosity in the offspring, causing severe morbidity or even mortality due to significant hemolysis. Awareness of these low expression alleles is important for genetic counseling purposes. Molecular studies, although becoming more mainstream, have not been used extensively to diagnose these disorders. This is most likely due to the complex genetic nature of these disorders (e.g. large genes with multiple exons involved, and multi-gene disorders (i.e. hyperbilirubinemia due to HS as well as involvement of genes involved in bilirubin metabolism). The accessibility of next generation sequencing (NGS) methods in the clinical laboratory has made diagnosing complex genetic disorders feasible.
Our current diagnostic panel includes 28 genes encoding cytoskeletal proteins and enzymes, and covers the complete coding region, splice site junctions, and, where appropriate, deep intronic or regulatory regions. Targeted gene capture and library construction for NGS are performed using a Sure Select kit (Agilent). Indexed samples are quantified using qPCR and then pooled prior to sequencing on the Illumina NextSeq or HiSeq instruments. Samples are sequenced using 150 bp paired-end sequencing. This panel includes genes responsible for RBC membrane defects, enzyme deficiencies, as well as bilirubin uridine diphosphate glucuronosyltransferase (UGT1A) genes that have a distinct role in hyperbilirubinemia.
We now report the first 268 patients evaluated using our NGS panel between 2015-2018. These patients were evaluated using an Institutional Review Board Protocol (IRB - 00077285). The age of the patients ranged from newborn to 68 years. These patients presented with symptoms ranging from mild lifelong anemia to severe hemolytic anemia with extreme hyperbilirubinemia. Genetic variants were classified according to the American College of Medical Genetics (ACMG) guidelines.
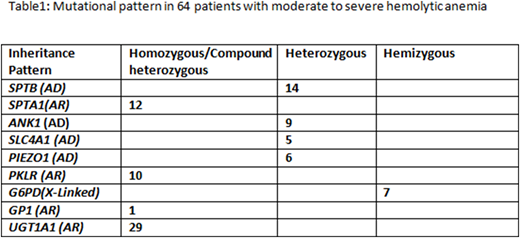
We identified pathogenic and likely pathogenic variants in 64/268 (24%) patients that were clearly responsible for the disease phenotype (e.g. moderate to severe hemolytic anemia). Approximately half of them were novel mutations. Moreover, 29/268 (11%) of patients were homozygous for a promoter polymorphism in the UGT1A1 gene A(TA)7TAA (UGT1A1*28), which may lead to reduced expression of the UGT1A1 gene and Gilbert's syndrome. Furthermore, 4/29 UGT1A1 polymorphism cases were associated with pathogenic spectrin mutations, likely increasing the severity of the clinical phenotype in these patients. Overall, the most commonly mutated genes were SPTB and SPTA1, encoding spectrin subunits, followed by PKLR and ANK1 (Table 1). Complex interactions between variants in the SPTA1 gene and the common alpha-LELY and alpha-LEPRA alleles were predicted to be associated with HPP and autosomal recessive HS in 12/64 patients. Furthermore, 23/268 (9%) patients had mutations that were predicted to cause moderate to severe anemia if inherited with another mutation, making them important for genetic counseling purposes (data not shown). Our results demonstrate that many patients with hemolytic anemia harbor complex combinations of known and novel mutations in RBC cytoskeleton/enzyme genes. Many variants of unknown significance were also identified that could potentially contribute to disease.
To conclude, the use of NGS provides a cost-effective and comprehensive method to assist in the diagnosis of hemolytic anemias, especially in instances where complex gene-gene interactions are suspected.
No relevant conflicts of interest to declare.

